Quantum-chemical calculations have been successfully used to complement the experimental X-ray powder diffraction (XRPD) data at several stages of the structure solution process:
- To optimize the molecular geometry in the gas phase to obtain accurate starting molecular structure suitable for structure solution by real space methods. In this stage different levels of theory are applied: molecular mechanics, semi-empirical methods, Hartree-Fock methods, density functional theory (DFT).
- To minimize the energy of crystal structure and provide reasonable bond lengths and angles to input into the Rietveld refinement as chemical restraints. Calculations are performed using plane wave DFT with dispersion correction (DFT-D).
- Because it is difficult to accurately locate the positions of H atoms from the XRPD data, energy optimization provides the most reasonable approach to compute optimal positions for the H atoms as the final step, with unit cell and non-H atomic positions fixed to those established by Rietveld refinement.
- To clarify an ambiguity concerning the orientation of functional group that could not be distinguished on the basis of the XRPD data alone.
- Geometry optimization with DFT-D approach in the solid state has been applied to refine the crystal structure when Rietveld refinement yielded inaccurate molecular geometry, providing results whose accuracy is comparable to that of single-crystal refinement.
- To assess the correctness of experimental organic crystal structures
EXPO2014 provides crystallographers with tools to perform theoretical calculations for the geometry optimization. The first geometry optimization should be done with a faster level of theory, such as molecular mechanics or a semiempirical method. Once a geometry close to the correct geometry has been obtained with this lower level of theory, it is used as the starting geometry for a second optimization at the final, more accurate level of theory (e.g., DFT).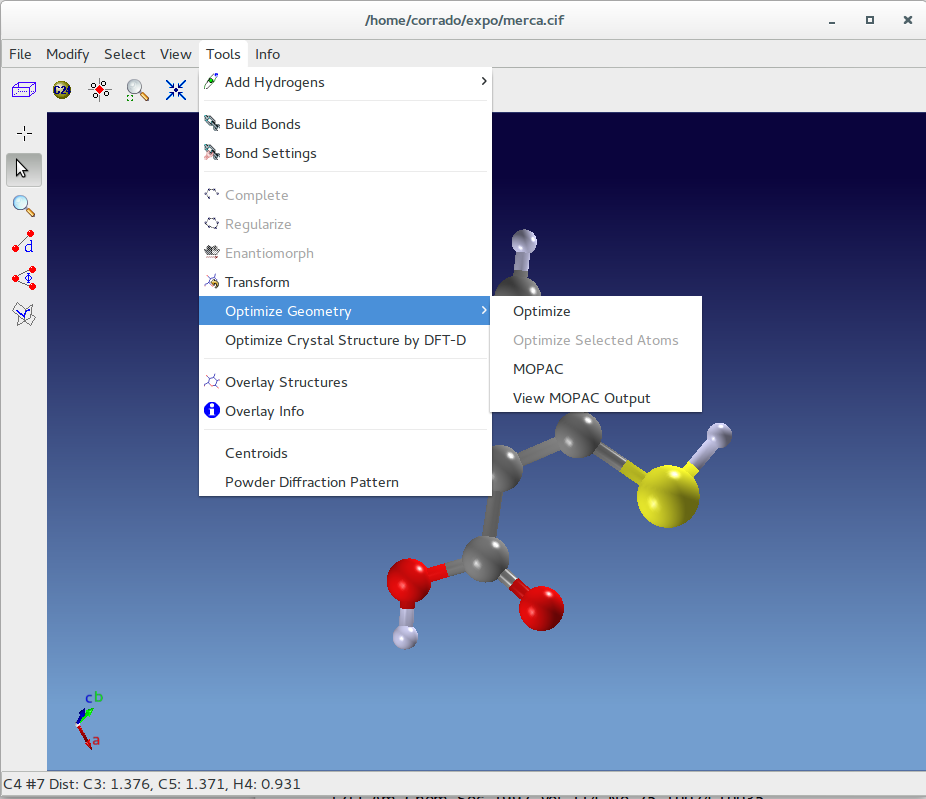
In molecular mechanics (MM), the energy of a compound consists of the sum of simple classical equations and the molecule is described as a collection of balls (corresponding to the atoms) held together by springs (corresponding to the bonds). The molecular mechanics model clearly does not use wave function and electrons are not explicitly included.
The total potential energy is typically taken to be the sum of the bond stretching energy Estr, the bending energy Ebend , the twisting (or torsion) energy Etor, and the energy of interaction between non-bonded atoms Enon-bond. The last contribution includes van der Waals, steric, and electrostatic interactions between atoms not chemically bound.
Etot = Estr + Ebend + Etor + Enon-bond
The equations for the potential energy terms contain parameters. The specified mathematical form of these equations and the parameters in them constitute a particular forcefield, and MM methods are sometimes called forcefield methods. The constants in this equation are obtained either from spectroscopic data or ab initio calculations. Many good force fields have already been developed and they differ in the number of terms in the energy expression, the complexity of those terms, and the way in which the constants were obtained.
To optimize the geometry and find the more stable conformation, each of the forces fi acting on a nucleus by electrons and other nuclei must vanish:
fi = – ∂Etot/∂qi
where qi are the nuclear coordinates of i-th atom.
In EXPO2014 two molecular mechanics force fields are used:
- The Merck molecular force field (MMFF) can be used for organic molecules and biomolecules [1]
- The Universal force field (UFF) is a full periodic table force field. This is widely used for system containing inorganic elements [2]
In EXPO2014 the support for molecular mechanics is provided using the class OBForceField from the library Open Babel [3]
Clicking on Tools > Optimize Geometry > Optimize MMFF94 force field will be applied and in case of failure (e.g. inorganic elements) UFF will be executed. Use Modify> Undo Optimize Geometry to restore the initial molecular geometry.
The geometry optimization can also be applied only on selected atoms:
- click on
 to activate the selection mode,
to activate the selection mode, - select by mouse atoms to apply the geometry optimization,
- click on Tools > Optimize Geometry > Optimize Selected Atoms.
E.g., you are interested to optimize only the position of H atoms, fixing the position of all other atoms: click on Select > Select Type > H atoms and then Tools > Optimize Geometry > Optimize Selected Atoms.
The most frequent use of MM is undoubtedly to obtain reasonable structures for other kinds of calculations: semi-empirical (SE), ab initio or density functional theory (DFT). The two salient features of MM calculations on small to medium-sized molecules is that they are fast and they can be very accurate. MM calculations might still take only seconds. MM, even when it does not provide very accurate geometries, can supply reasonably good input geometries for SE, ab initio or DFT, and this is one of its main applications.
There are two general approaches to solving the Schrödinger equation of a molecular system: semi-empirical and ab initio methods. The semi-empirical methods assume an approximate Hamiltonian operator and the calculations are further simplified by approximating integrals with various experimental data such as ionization energies, electronic spectral transition energies, and bond energies. On the other hand, ab initio methods use a “correct” Hamiltonian operator, which includes kinetic energy of the electrons, attractions between electrons and nuclei, and repulsions between electrons and those between nuclei, to calculate all integrals without making use of any experimental data other than the values of the fundamental constants. An example of these methods is the self-consistent field (SCF) method first introduced by D. R. Hartree and V. Fock in the 1920s.
MOPAC (Molecular Orbital PACkage) is one of the most widely used semi-empirical software packages and is designed for a wide range of functionality. We tested version 2012 and 2016. MOPAC is not distributed with EXPO, so a copy of the software needs to be separately downloaded. For more information or to obtain a copy, please visit http://www.openmopac.net/MOPAC2016.html, the software is currently free for academic use.
An interface of MOPAC is provided in EXPO and allows users to perform a gas-phase geometry optimization. This approach is useful to generate molecular model with accurate angles and bond lengths, suitable as a starting model for structure solution by real space methods. The advantage of semi-empirical calculations is that they are much faster than SCF or DFT methods and can be applied routinely to large system.
A MOPAC optimization is performed selecting Tools > Optimize Geometry > MOPAC from the principal menu.
The calculation is performed by choosing the following options in the MOPAC dialogue window: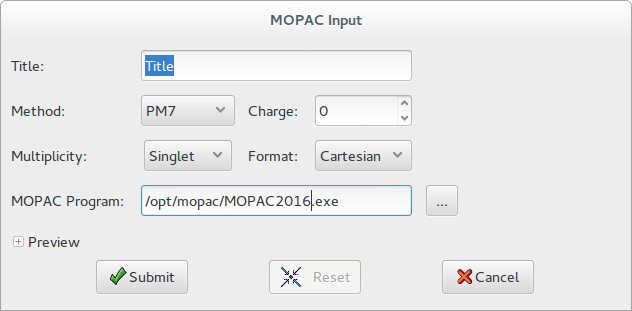
Title: define the title of the calculations, e.g., the name of molecule. It is reported in the second line of the MOPAC input file
Method: Different semi-empirical Hamiltonians are available and are used in the electronic part of the calculation to obtain molecular orbitals and its derivative with respect to molecular geometry. Not all chemical elements are supported by the selected Hamiltonian. For more information about the elements available for the specified method see the geometry section in MOPAC manual: http://openmopac.net/manual/
Charge: Write the charge of studied system
Multiplicity: Allow the user to specify the number of singly occupied orbitals. Singlet is the default, and specify a closed shell that includes the most of organic molecules; doublet specify one singly occupied orbital (e.g. organic radicals); triplet specifies two singly occupied orbitals; and so forth. If the multiplicity is unknown, you have to try all.
Format: Geometries can be specified within MOPAC using the Cartesian coordinate definition or the Gaussian Z-matrix format. The position of molecule in the cell unit will be lost if the geometry is defined in Z-matrix format.
Mopac Program: EXPO2014 automatically locates the position where MOPAC exists, if MOPAC is installed in a standard directory (C:/Program Files/MOPAC/ on Windows, /opt/mopac/ on Linux) or the environmental variable PATH includes the installation directory, otherwise the MOPAC executable must be specified directly by the user.
If you click on Submit, MOPAC will be launched and at the end of the geometry optimization, the new geometry will be displayed and an output file, called structure_name_mopac.out, will be produced in the working directory. The output file can be viewed from Tools> Optimize Geometry> View Mopac Output. You can read previously generated MOPAC output files and display the molecular geometry from File > Import Structure and selecting ‘MOPAC Output File (*.out)’ in the file extension menu.
Before performing the calculation, add the missing hydrogen. You can use the option Tools>Add Hydrogens. Wrong starting geometry, missing hydrogens or wrong multiplicity are possible causes of an incorrect result. In this situation use Modify> Undo Optimize Geometry to restore the initial geometry, then read the output file to find a possible cause of error.
Additional calculations can be performed by clicking the preview button and changing the input file. For example, it might be particularly important to calculate the bond order: add the keyword BONDS in the first line and bond order between all pairs of atoms will be printed in the output file. The keyword NOOPT can be used to perform only an energy calculation; the calculated energy will be written in the output file. Use the keyword NOOPT OPT-H to optimize the positions of hydrogen atoms while freezing the positions of all other atoms. A complete list of keywords used in MOPAC2016 is available at link http://openmopac.net/manual/allkeys.html
Another approach closely related to the ab initio methods that has gained increasing prominence in recent years is the DFT. This method bypasses the determination of the wavefunction ψ. Instead, it determines the molecular electronic probability density ρ directly and then calculates the energy of the system from ρ.
EXPO2014 creates input files for well known quantum chemistry programs, clicking on File > Export Structure. Input files for GAMESS [4] and NWCHEM [5] contain keywords for geometry optimization by density functional theory (DFT) calculations to perform using the B3LYP functional and a standard 6-31G* basis set.
In addition Tools > Optimize Crystal Structure by DFT-D can create an NWCHEM input file for the optimization of crystal structure by plane wave density function theory with Grimme dispersion correction. In order to obtain a good accuracy on the interatomic distances, you should do a convergence test with respect the cutoff energy and a convergence study associated with the sampling of the Brillouin zone.
At the end of the DFT-optimization, the output file generated by the quantum chemistry software can be imported in EXPO2014 and the calculated crystal structure can be visually compared and superimposed with the experimental one by using Tools > Overlay Structures to generate an overlaid.
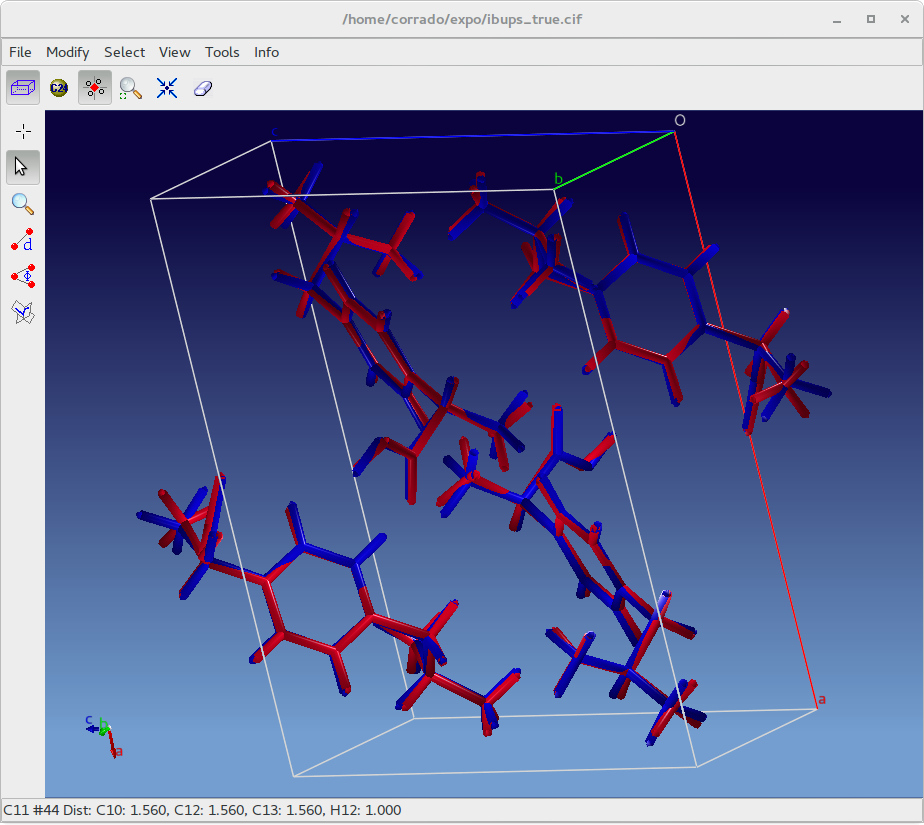
The quality of the agreement between the experimental and the theoretical structure can be evaluated in terms of root mean square displacement (RMSD) calculated for all non-H atoms. In a recent paper 215 organic crystal structures determined from X-ray powder diffraction data and published in an IUCr journal were energy-minimized with DFT-D and compared to the single crystal structure (van de Streek & Neumann, 2014). According to this survey the value of RMSD never exceeds 0.35 Å for correct crystal structure. RMSD information is also available in EXPO2014 by Tools > Overlay info.
[1] Thomas A. Halgren, J. Comput. Chem., 17, 490-519 (1996)
[2] J. Am. Chem. Soc. 1992, Vol. 114, No. 25, 10024-10035
[3] N M O’Boyle, M Banck, C A James, C Morley, T Vandermeersch, and G R Hutchison. “Open Babel: An open chemical toolbox.” J. Cheminf. (2011), 3, 33.
[4] http://www.msg.ameslab.gov/gamess/
[5] http://www.nwchem-sw.org/index.php/Main_Page
van de Streek, J. and Neumann, M.A. “Validation of molecular crystal structures from powder diffraction data with dispersion-corrected density functional theory (DFT-D)” Acta Cryst. (2014). B70, 1020–1032.