The sub-menu File is shown.
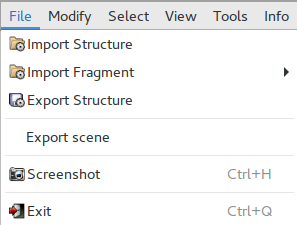
It allows:
|
Load a structure from file |
|
|
Load a molecular fragment from file |
|
|
Export current crystal data to a file |
|
|
Export Scene |
Export STL file for 3D printing |
|
Produce an image file of Jav screen |
|
|
Close the viewer |
Import
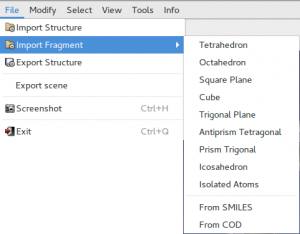
It is possible to read crystal structure from the following types of file:
- Crystallographic Information File (*.cif) http://www.iucr.org/resources/cif
- MDL Molfile (*.mol) http://accelrys.com/products/informatics/cheminformatics/ctfile-formats/no-fee.php
- MOPAC File Formats. It can handle data structure in Cartesian coordinates or in internal coordinates. http://openmopac.net/manual/index.html
- Tripos SYBYL Mol2 File (*.mol2, *.ml2) http://tripos.com/data/support/mol2.pdf
- Protein Data Bata Bank (*.pdb) http://www.wwpdb.org/
- Fenske-Hall Z-matrix (*.zmt), Z-matrix file created by Open Babel
- Free Form Fractional (*.fra, *.frac) http://openbabel.org/wiki/Free_Form_Fractional
- XYZ Cartesian Format (*.xyz) http://en.wikipedia.org/wiki/XYZ_file_format
- Input/Output files of SHELXL (*.ins,*.res) http://shelx.uni-ac.gwdg.de/SHELX/
EXPO2014 can also read the coordinates of the final molecular geometry in the output files generated by well known quantum chemistry programs:
- GAMESS output file (*.out,*.log) http://www.msg.ameslab.gov/gamess/
- NWCHEM output file (*.out,*.log) http://www.nwchem-sw.org/index.php/Main_Page
- MOPAC output file (*.out) http://openmopac.net/MOPAC2016.html
- ABINIT output file (*.out) http://www.abinit.org/
- CRYSTAL output file (*.out) http://www.crystal.unito.it/index.php
- Quantum ESPRESSO output file (*.out) https://www.quantum-espresso.org/
In default the program export in the external file the asymmetric unit.
Isolated atoms and some common chemical geometries (tetrahedron, octahedron, square plane) can also imported. They are useful building blocks in structure solution by global optimization.
The program can read molecule from specified SMILES string and has the ability to import CIF files by querying the Crystallographic Open Database (COD). The button From COD allows the access to a graphical interface useful to build query to COD from appropriate search parameters (text contained in the metadata of the entry, chemical formula of the crystal, type and number of chemical element symbols). HTTP requests to get data from COD are performed by using RESTful API made available by COD developers to facilitate searches in the COD.
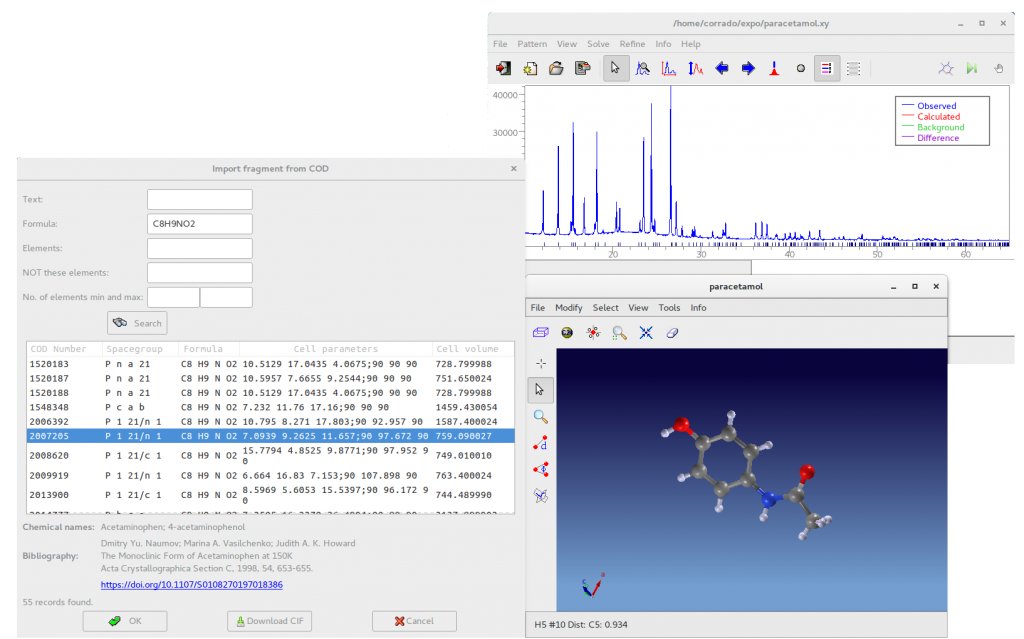
The picture above shows the result of the query to COD searching for the chemical formula of paracetamol (C8H9NO2). All the molecules having the requested formula are listed and the user can visualize the 3D structure by selecting a row in the list. If powder data has been previously loaded, the program computes the reflections of the selected structure and displays them below the pattern in the main window, allowing a visual match between the experimental pattern and the reflections calculated from structure in the COD. Before the query, if the JAV viewer is closed, select from the menubar of the main window View > JAV Molecular Viewer.
Export
Information about the currently displayed crystal structure can be exported in the following files:
- Crystallographic Information File (*.cif) http://www.iucr.org/resources/cif
- Input/Output files of SHELXL (*.ins,*.res) http://shelx.uni-ac.gwdg.de/SHELX/
- Free Form Fractional (*.fra, *.frac) http://openbabel.org/wiki/Free_Form_Fractional
- Protein Data Bata Bank (*.pdb) http://www.wwpdb.org/
- POV-Ray input file (*.pov) input file for the freeware program POV-Ray. http://www.povray.org/download/
- MDL Molfile (*.mol) http://accelrys.com/products/informatics/cheminformatics/ctfile-formats/no-fee.php
- Fenske-Hall Z-matrix (*.zmt), Z-matrix file created by Open Babel
- MDL Molfile (*.mol) http://accelrys.com/products/informatics/cheminformatics/ctfile-formats/no-fee.php
- XYZ Cartesian Format (*.xyz) http://en.wikipedia.org/wiki/XYZ_file_format
- MOPAC File Formats with cartesian coordinates. http://openmopac.net/manual/index.html
EXPO2014 create input files for well known quantum chemistry programs. Input files for GAMESS and NWCHEM contain keywords for geometry optimization by density functional theory (DFT) calculations to perform using the B3LYP functional and a standard 6-31G* basis set. This approach is useful to generate molecular model with accurate angles and bond lengths, suitable as a starting model for structure solution by real space methods
- GAMESS input file (*.inp,*.gamin) http://www.msg.ameslab.gov/gamess/
- NWCHEM input file (*.out,*.log) http://www.nwchem-sw.org/index.php/Main_Page
- GAUSSIAN Cartesian Input (*.com, *.gau) http://www.gaussian.com/
- GAUSSIAN Z-matrix Input (*.gzmat) http://www.gaussian.com/
- ABINIT input file (*.in) http://www.abinit.org/
- CRYSTAL input file (*.d12) http://www.crystal.unito.it/index.php
- Quantum ESPRESSO input file (*.in) https://www.quantum-espresso.org/
In default, the program exports only the coordinates of the atoms in the asymmetric unit to a file. To export all displayed atoms select the ‘Displayed atoms’ option in the file selection dialog box.
Screenshot
Images of crystal structure displayed in the graphic area can be saved in files of different following formats:
- Bitmap File (*.bmp)
- ICO file (*.ico)
- jpeg file (*.jpg)
- Portable Network Graphics file (*.png)
- Tagged Image File (*.tif)
Check the option ‘Transparent’ in the file selection dialog box the obtain images with transparent background.
Exit
This is used to exit the program.