This tutorial provides a complete walkthrough of the ab initio crystal structure solution process for cimetidine, from indexing to the structure solution using direct methods.

The only required information by EXPO is the experimental powder diffraction pattern and the chemical formula C10H16N6S.
The examples directory contains all the necessary files:
- cime.exp: input file for EXPO
- cime.dat: a synchrotron X-ray diffraction data file, with the first line specifying the initial 2θ value, 2θ step size, and final 2θ value, followed by the experimental diffraction counts listed on the subsequent lines.
To access to examples directory:
- Open EXPO by double clicking on Expo icon.
- Click on File > Load Examples
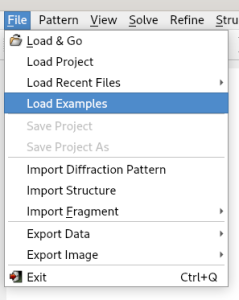
Then select the file cime.exp.
This file can be viewed and, if necessary, modified using the Edit button.
The contents of the file are shown below.
%structure cime %job cimetidine (C10H16N6S) -- Synchrotron data %data pattern cime.dat wavelength 1.52904 synchrotron %ntreor %continue
It contains the command (%data) for providing the minimal required information (name of data file, wavelength and type of radiation used to collect data, if different from laboratory X-rays) and the commands for activating the indexing process (%ntreor) and the ab-initio structure solution (%continue).
More information about the creation of input file can be found in the section Preparing input file.
Close the cime.exp file, and then click Open. If the output file (typically cime.out) already exists, a warning message will be displayed.
The powder diffraction data will then be shown. By selecting Next, an automatic peak search is performed. The d-values corresponding to the detected experimental peaks are then automatically provided to the indexing process.
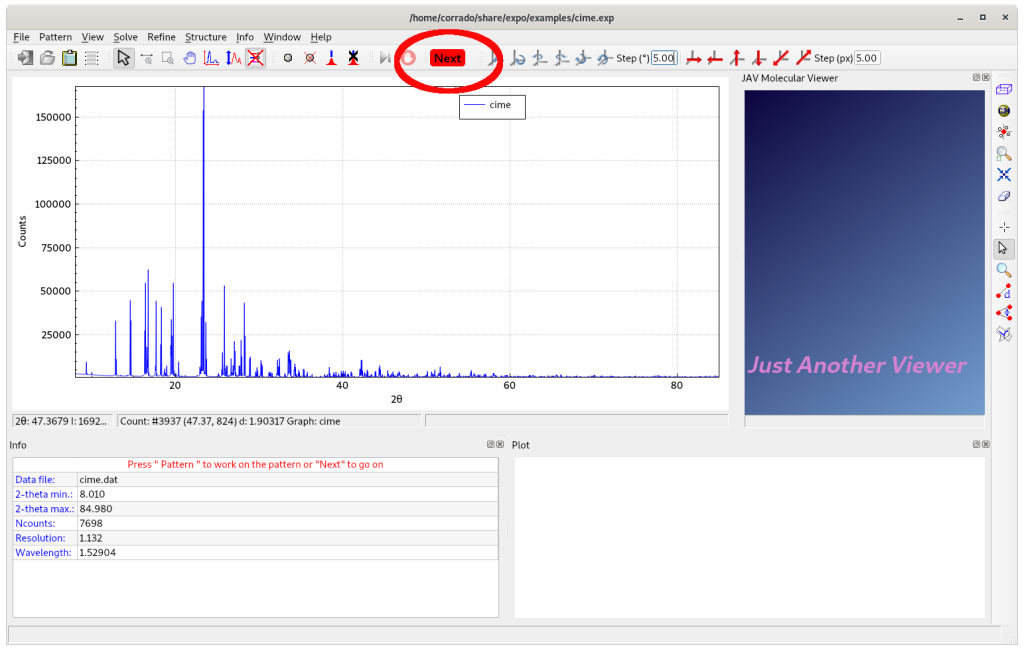
Upon completion, EXPO displays the indexing results through a graphical interface.
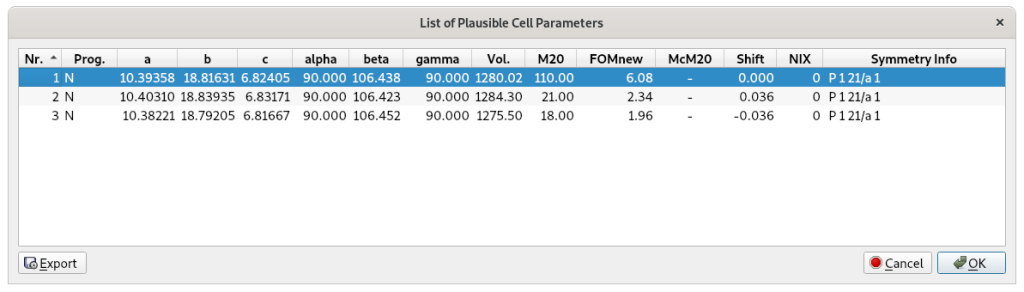
Click on OK to accept the selected plausible cell parameters and proceed to the next step: space group determination.
The space group determination step, as well as the application of direct methods, requires information about the unit cell contents. This can be easily estimated using the molecular formula and the cell volume (Vcell) which EXPO calculates from the unit cell parameters. The cell volume is displayed graphically and/or included in the EXPO output file.
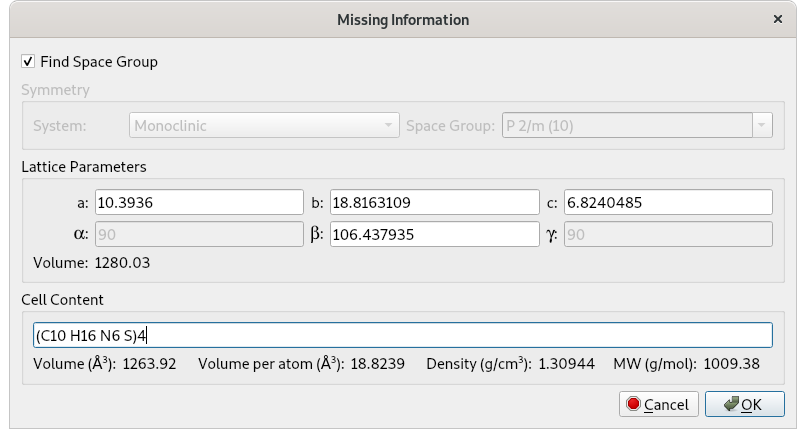
The cell content is obtained by multiplying Z by the molecular formula. You can calculate Z as the ratio between the cell volume (Vcell) and the molecular volume (Vmol): Z = Vcell/Vmol.
Vmol can be calculated by one of the two following alternative ways:
- Using the 18 Å3 rule: count all the non-hydrogen atoms in the chemical formula and multiply this number by 18 Å3;
- Using approximate atomic volumes: 14 Å3 for C, 12 Å3 for N, 11 Å3 for O, 5 Å3 for H, 26 Å3 for Cl, 25 Å3 for S.
For the cimetidine compound, the chemical formula is C10H16N6S and the cell volume is 1280 Å3. If the first criterion is applied, the molecular volume is given by: Vmol = 18 x 17 = 306 Å3. Using the second approach Vmol = 14 x 10 + 5 x 16 + 12 x 6 + 26 x 1 = 318 Å3.
The corresponding Z value is: Z = Vcell/Vmol = 1280/306 = 4.18, which can be approximated to 4. Therefore, 4 molecules of cimetidine are present in the unit cell, and the unit cell content to be supplied to EXPO via graphic interface is either (C10 H16 N6 S)4 or C40 H64 N24 S4. A valid molecular formula for the unit cell content must meet the same criteria as those adopted for string in the directive content of the command %data.
Click on OK to perform the space group determination, then click the Next button to go on. At the end of the procedure, a list of the space groups and extinction symbols, compatible with the lattice symmetry and ranked according probability values, will be displayed:
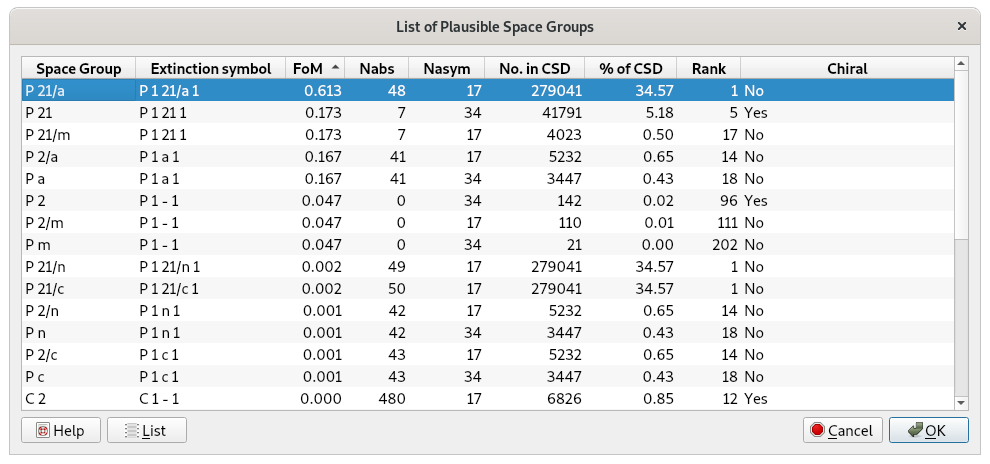
The most plausible extinction symbol P 1 21/a 1, is automatically selected. By clicking OK, the corresponding space group, P 21/a, is applied. EXPO asks whether to transform P 21/a into the standard setting P 21/c. We can continue the solution process with P 21/a by keeping the No button selected.
After the determination of the space group, EXPO proceeds to the extraction step, which is performed using the selected space group. Continue by clicking the Next button. At the end of the process, a list of integrated intensity values, extracted from the experimental powder pattern, is generated and automatically passed to the Direct Methods process for structure solution.
The final result is visualized on JAV Molecular Viewer.
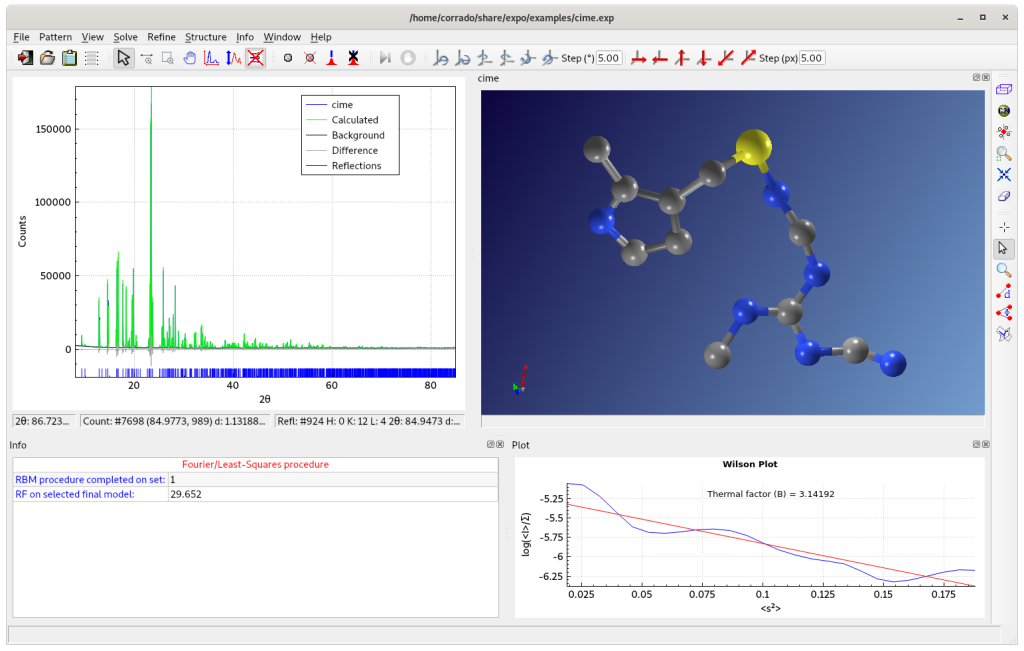
If errors in the assignment of carbon or nitrogen atomic species are identified in the final structure model, they can be corrected using graphical options from the Structure > Modify menu. Hydrogen atoms can be geometrically located from Structure > Tools > Add Hydrogens.
The aim of this example is to guide the user in the event of a default EXPO run failure using direct
methods.
To access the files needed for this test:
1. Open EXPO by double-clicking the EXPO icon.
2. Click File > Load Examples.
3. Select the input file merca.exp. This file can be viewed and, if necessary, modified using the Edit button.
%structure merca %job 2-Mercaptobenzoic acid %data cell 7.885 5.976 14.949 90.0 100.48 90 spacegroup p 21/c content (C7 O2 S1 H6)4 pattern merca.xy wavelength 1.54056 %continue
In this example, it is assumed that the cell and space group have already been determined, and the unit cell contains 4 molecules of C7H6O2S.
Click on the Next button to go on continuously.
The final structure model generated by EXPO may not accurately represent the correct crystal structure of 2-Mercaptobenzoic acid, as it is not chemically interpretable.
In a default run of EXPO, direct methods generate several phasing trials. No more than 20 of these trials are stored and ranked in decreasing order based on a suitable combined figure of merit (CFOM). The phase set with the highest CFOM is automatically selected for structure solution. If this fails to yield the correct structure, the ALLTRIALS procedure can be applied, which explores all remaining stored phasing trials in search of a correct solution.
This can be done automatically via the graphical interface by:
a) Selecting Solve from the top-level menu, then choosing Explore Trials.
b) Checking the Explore trials not processed yet option.
c) Clicking on Explore.
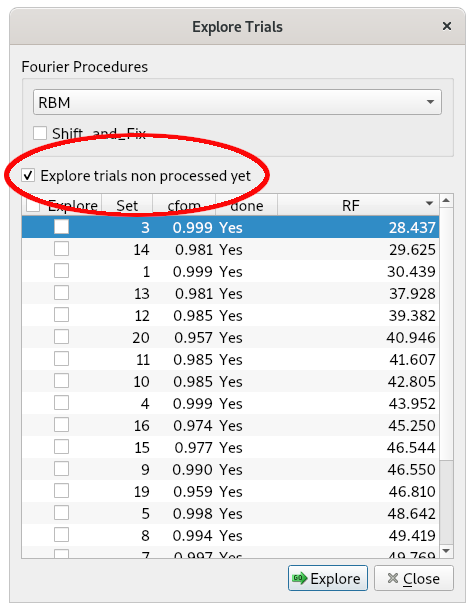
At the end of the procedure, EXPO ranks the phasing trials (and the corresponding structure models) in order of increasing RF agreement factor values.
The best solution, corresponding to the lowest RF value, is selected and displayed. Close the ‘Explore Trials’ window. In the selected model, some chemical labels may be incorrect and should be corrected.
The purpose of this tutorial is to guide the user through the crystal structure solution process of the paracetamol compound (form I polymorph) using direct space methods, particularly the simulated annealing process.
To access to examples directory:
- Open EXPO by double clicking on Expo icon.
- Click on File > Load Examples
Then select the input file paracetamol.exp.
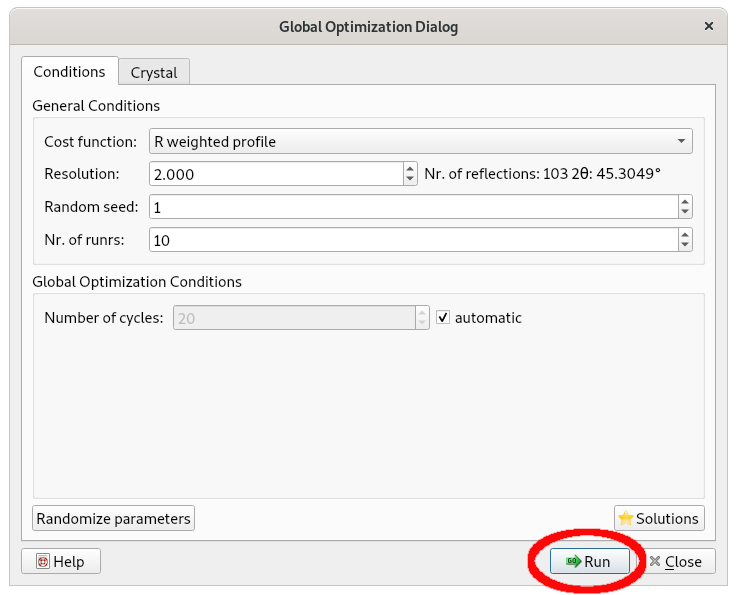
By clicking the Open button, the file paracetamol.mol is loaded and the initial model for the simulated annealing optimization will be displayed and the following window will appear. By selecting Run, the simulated annealing procedure performs multiple runs, 10 by default.
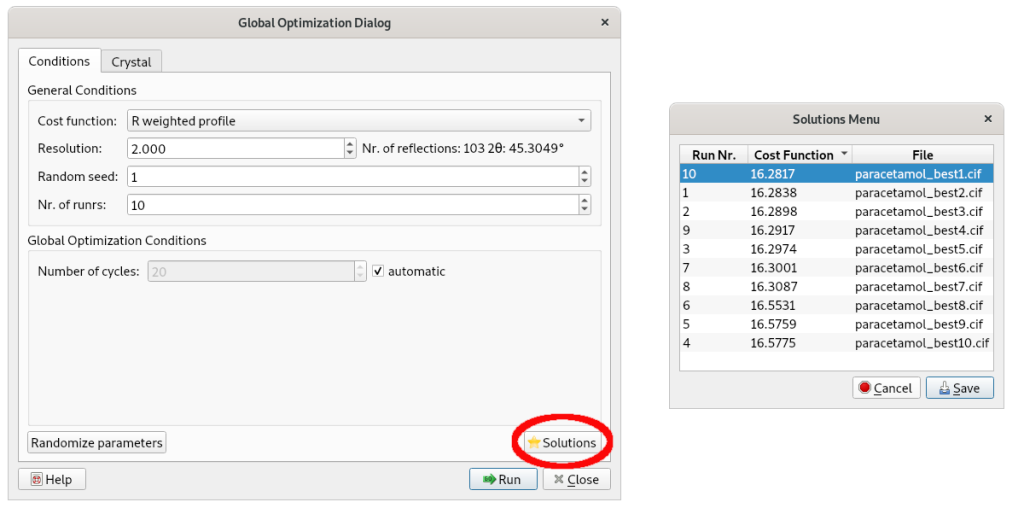
The structure models corresponding to each simulated annealing run can be explored using the Solutions button.
These models are ranked according to their cost function values and can be visualized and analyzed. CIF files corresponding to each structure model are generated.
Click Save, to select one of the simulated annealing solutions.
Cimetidine – Cernik, R.J., Cheetham, A.K., Prout, C.K., Watkin, D.J., Wilkinson, A.P., Willis, B.T.M. (1991). J. Appl. Cryst. 24, 222-226.
2-Mercaptobenzoic acid – Steiner, T. (2000). Acta Cryst. C56, 876–877.
Paracetamol (form I polymorph) – Nichols, C. & Frampton, C. S. (1998). J. Pharm. Sci.87, 684–693.