Molecular Replacement (MR) is the most popular method for solving protein crystal structures. Sir2019 uses an updated version of REMO09 (Caliandro et al., 2009) for orienting and locating, in the unit cell of the target structure, the various monomers constituting the model. REMO09 breaks the six dimensional search into 3-dimensional rotation and translation steps; for both of them the program automatically decides the working resolution. The program may be schematized in the following three steps:
-
- the space group of the model structure is assumed to be the symmorphic variant of the protein space group;
- fitting between model and target structure is searched in reciprocal space by rotating the reciprocal lattice of the protein with respect to the calculated transform of the model structure;
- the oriented model molecule is located in the target unit cell by using correlation functions calculated by Fast Fourier Transform.
The approach is purely probabilistic: the various figures of merit are all derived by means of the method of joint probability distribution functions.
Rotations and translation parameters are refined via simplex method. The phase values so obtained are extended via a modified version of the pipeline REVAN (Carrozzini et al., 2015), combining REMO09, REFMAC, REFMAC phase driven, DM, VLD, FL and BUCCANEER (Cowtan, 2006), as default choice; if other Automated Model Building (AMB) programs are available (Nautilus, ARP/wARP or Phenix) the program can use them in an automatic way in order to supply a refined and interpreted solution.. Models with sequence similarity index below 0.40 are automatically submitted to mutations, to make the model more similar to the target. The only permitted mutation is the trivial truncation of the residues to alanine. As in the standard REVAN version, when data resolution is smaller than 1.25 Å, LDT is preferred to DM because of its larger effectiveness. The modified model is cyclically resubmitted to REFMAC phase driven- EDM-VLD-FL cycles.
The AMB program is used not only to check if the resulting model may be automatically interpreted, but also as intermediate step for improving phases which are resubmitted to cycle of EDM-VLD-FL.
It is possible to create the input file for Sir2019 using a text editor; as an example:

The list of directives is available at the end of this page or here. The directives for %data in case of proteins, are available here
A graphic interface is available in Sir2019 to work with Molecular Replacement by means of the New item ![]() in the toolbar or in the menu File :
in the toolbar or in the menu File :
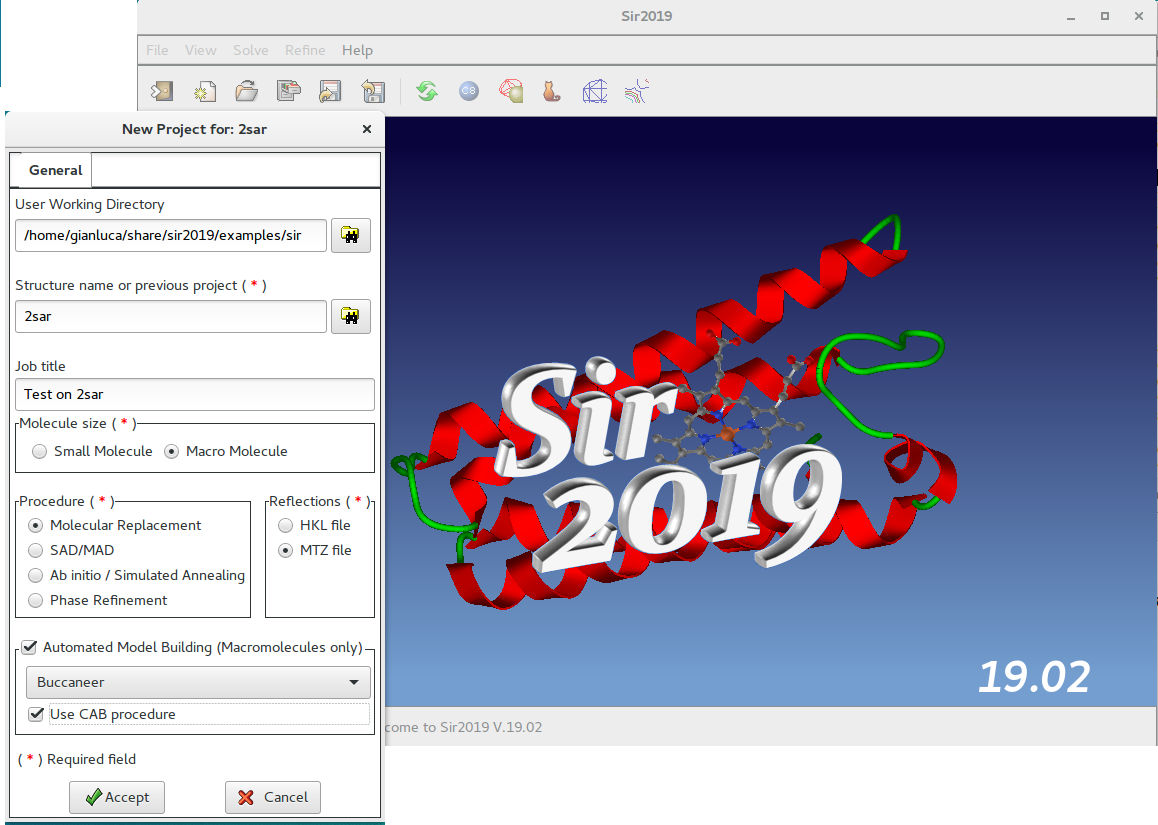
In this window the user should specify, among others, the working directory, the molecule size (small or macro), the procedure to use (Molecular Replacement) and the reflection file format (MTZ).
Proteins only: if an Automated Model Building program is installed, it is possible to use it by selecting Buccaneer or Nautilus or ARP/wARP or Phenix. If selected the CAB procedure will be applied at the end of the Molecular Replacement.
Once clicked on ![]() it is possible to access the MTZ section:
it is possible to access the MTZ section:
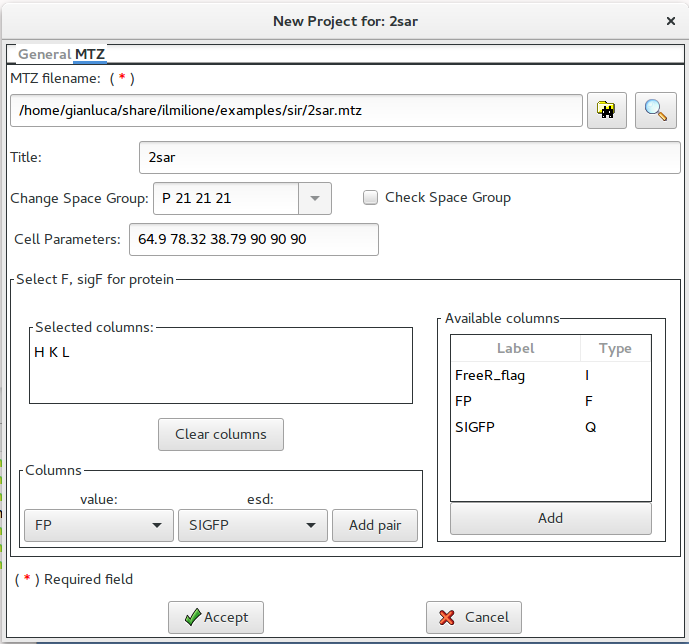
Using this window it is possible to supply the name of the MTZ file and to select the necessary columns (H K L FP SIGFP).
Once clicked on ![]() it is possible to access the Cell Content section:
it is possible to access the Cell Content section:
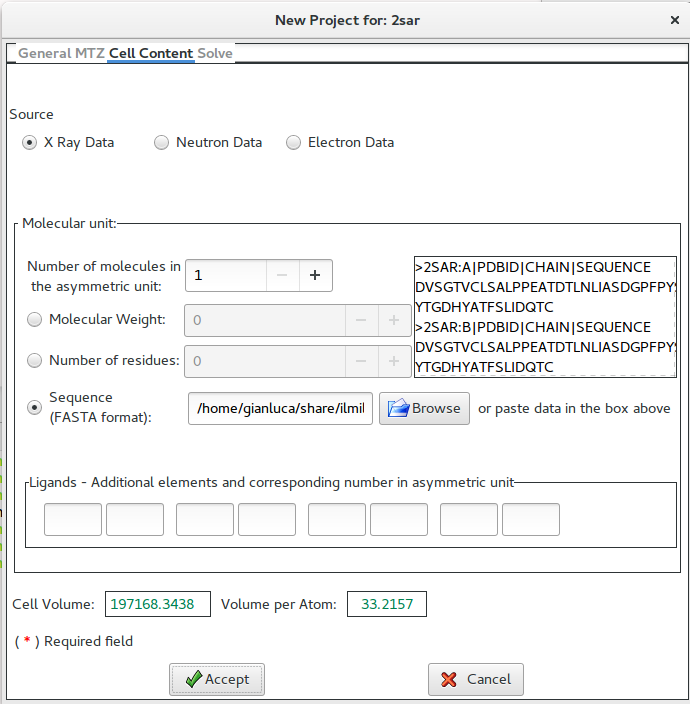
In this example, the sequence (in FASTA format) has been supplied.
After these data have been accepted, the Solve interface is available:
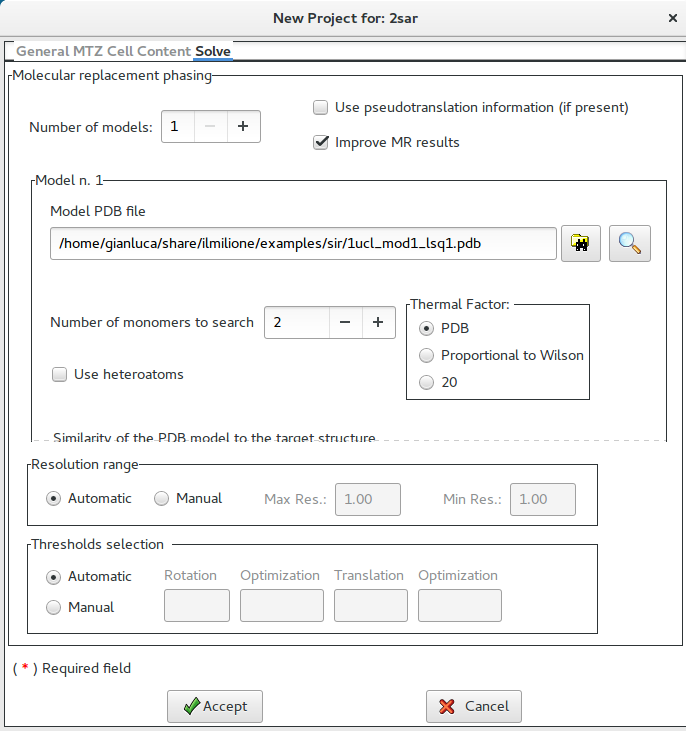
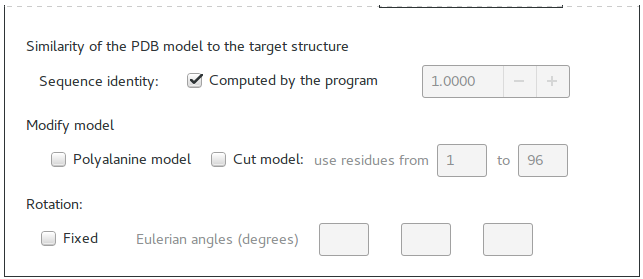
It is possible to specify the Sequence identity (otherwise it is computed by the program).
The model can be modified transforming it in a sequence of alanine residues or it can be cutted.
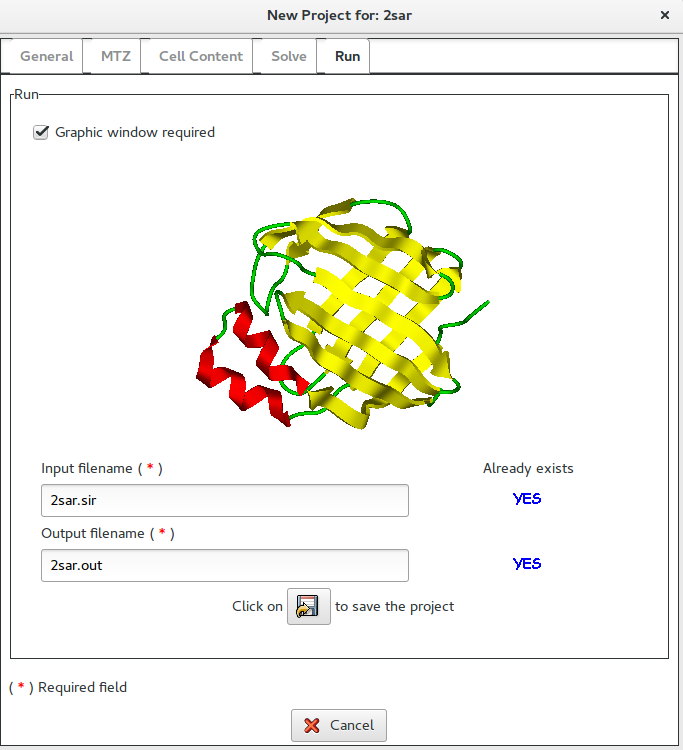
Once clicked on ![]() it is possible to access the Runsection:
it is possible to access the Runsection:
If Graphic window required is checked, a graphical output will be produced (see below).
In this section it is possible to change the default names for the input (project) and for the output file. The user, by clicking on ![]() button, can save the input file for Sir2019.
button, can save the input file for Sir2019.
Now it is possible to edit ![]() the input file (2sar.sir); to start calculations click on
the input file (2sar.sir); to start calculations click on ![]() .
.
Once the program is over, by means of the “View output file” ![]() feature in “File” it is possible to access the complete output file.
feature in “File” it is possible to access the complete output file.
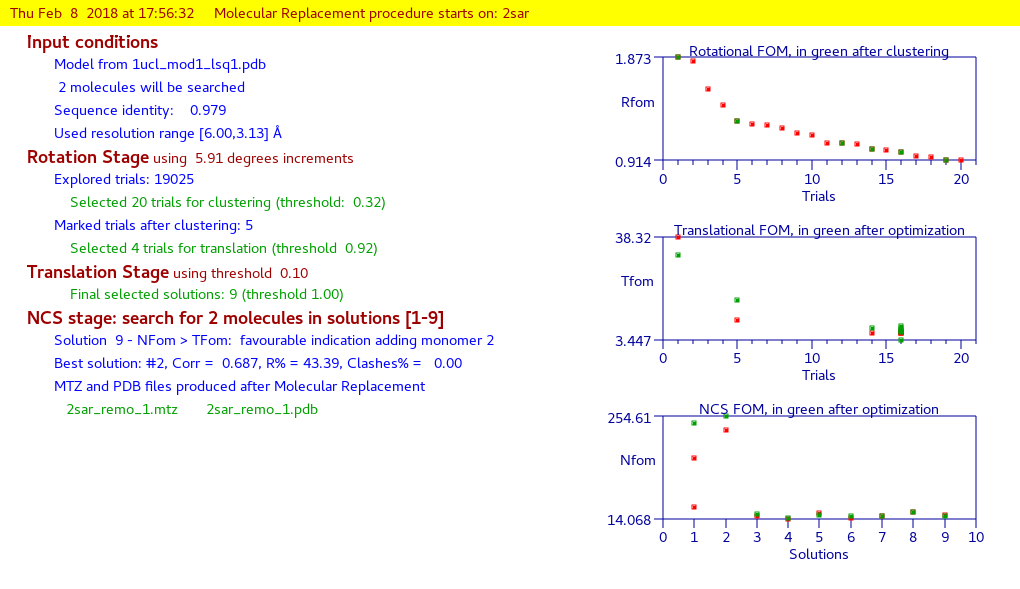
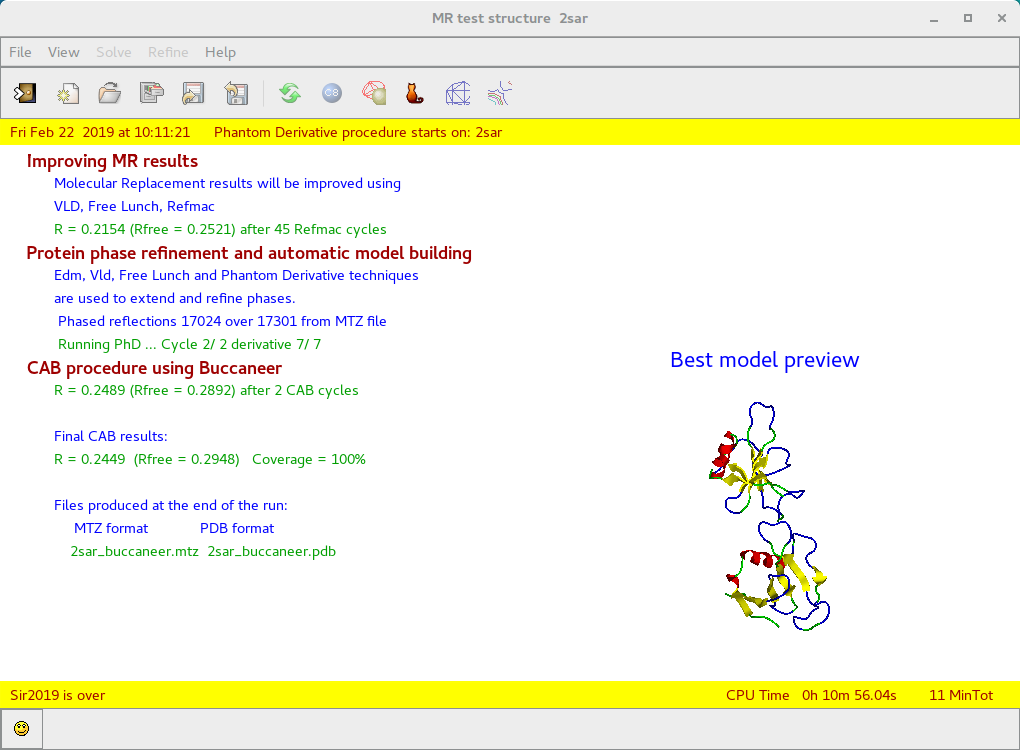
By means of the “Summary” ![]() feature in “File” menu is possible to get a synthetic output and also the graphic windows produced by the program during the run (in HTML format). An example of graphical output follows.
feature in “File” menu is possible to get a synthetic output and also the graphic windows produced by the program during the run (in HTML format). An example of graphical output follows.
First part:
application of Moleculare Replacement procedure
Second part:
improving MR results, protein phase refinement and CAB procedure application using Buccaneer.
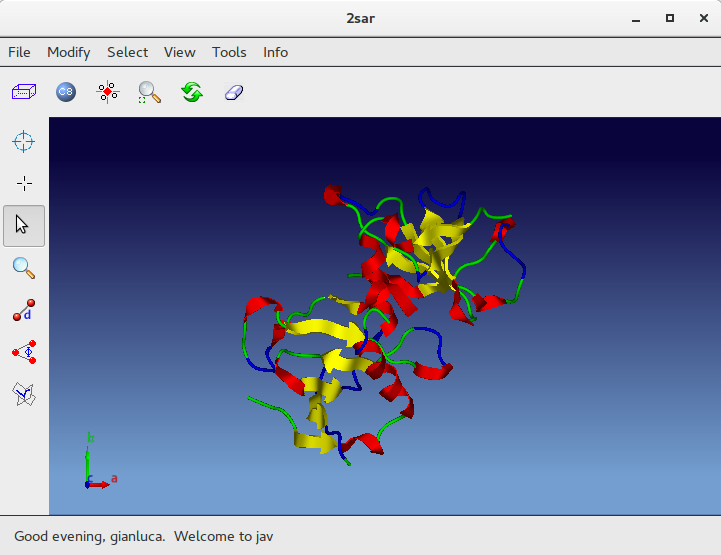
When the program is over it is possible to interact with the structure using ![]() Jav:
Jav:
PSEUDO
The pseudotranslational information will be actively used to solve the protein structure.
APPROT
The program just finds the orientation of a model and stops.
SHELX
The program export the output coordinates files in SHELX format (PDB format is the default).
MODEL
Starts the block of directives related to each model.
ENDMODEL
Ends the block of directives related to each model.
FRAGMENT [HETAM] string
String is the name of the coordinates file (*.pdb). HETATM allows the program to use Heterogenic atoms content.
IMPROVE
The MR model will be improved using VLD and, if the resolution is worse than 1.25 Å, by means of the Free Lunch algorithm.
REFINE n
The MR model will be refined by Refmac (if installed) using batches on n refinement cycles. The default is REFINE 10.
NOREFINE
No refinement by Refmac of the MR model.
NOFREELUNCH
The Free Lunch algorithm will not be used.
ALIGNMENT string
String is the name of the file (in FASTA format) containing the alignment between the sequence of the target and the sequence of the model.
POLY
The polyalanine model will be applied.
BFACTOR x / wilson / pdb
The atomic B factors are set to x or to the Wilson B-factor or to the PDB values.
CUT n1 n2
The program removes from the model some terminal residues if they lie ‘outside’ n1 and n2 limits.
RANGE x y
Resolution range (in Å) the program will use. If only one limit is given, it is the high resolution limit; otherwise must be x < y.
THRESHOLD x y z w
The program selects the solutions to be analyzed after the rotation, the clustering, the translation and the optimization stage; the cutoff value is the maximum normalized distance from the best solution FOM. The default threshold values are set automatically by the program and are shown on the graphic window and in the output file.
MONOMERS n
The number of monomer to search is set to n.
IDSEQUENCY x
Sequence identity with the target sequence (between 0.0 and 1.0). In default it will be calculated by the program.
POSITION r1 r2 r3 t1 t2 t3
The rotation (r1 , r2 , r3 eulerian angles) and the translation (t1 , t2 , t3 fractional values) are applied to the model.
ROTSELECTED r1 r2 r3
The rotation (r1 , r2 , r3 eulerian angles) is applied to the model; the procedure continues normally.
TRASELECTED t1 t2 t3
The translation (t1 , t2 , t3 fractional values) is applied to the model; the procedure continues normally.